

用分子模拟手段研究生物功能
Subtopic II. Understand the functions of biological molecules/systems or the mechanisms of chemical biological events through molecular modeling
J. Chem. Inf. Model. 2009, 49, 1033-1048.
Interpretation of the Binding Affinities of PTP1B Inhibitors with the MM-GB/SA Method and the X-Score Scoring Function
Xinglong Zhang, Xun Li, and Renxiao Wang*
We have studied the binding affinities of a set of 45 small-molecule inhibitors to protein tyrosine phosphatase 1B (PTP1B) through computational approaches. All of these compounds share a common oxalylamino benzoic acid (OBA) moiety. The complex structure of each compound was modeled by using the GOLD program plus the ASP scoring function. Each complex structure was then subjected to a molecular dynamics (MD) simulation of 2 ns long by using the AMBER program. Based on the configurational ensembles retrieved from MD trajectories, both MM-GB/SA and MM-PB/SA were employed to compute the binding free energies of all 45 PTP1B inhibitors. The correlation coefficient between the MM-GB/SA results and experimental binding data was 0.87 and the standard deviation was 0.60 kcal/mol. The performance of MM-PB/SA was slightly inferior to that of MM-GB/SA. Several aspects of the MM-GB(PB)/SA method were explored in our study to obtain optimized results. The X-Score scoring function was found to produce equally good results as MM-GB/SA on both the complex structures prepared by molecular docking and the configurational ensembles obtained through lengthy MD simulations. The structure-activity relationship of this set of compounds is also discussed based on the computed results. The computational approaches validated in our study are hopefully applicable to the study of other classes of PTP1B inhibitors.

J. Mol. Model. 2008, 15, 53-65
Molecular modeling of the three-dimensional structure of GLP-1R and its interactions with several agonists
Fu Lin and Renxiao Wang*
Glucagon-like peptide-1 receptor (GLP-1R) is a promising molecular target for developing drugs treating type 2 diabetes. We have predicted the complete threedimensional structure of GLP-1R and the binding modes of several GLP-1R agonists, including GLP-1, Boc5, and Cpd1, through a combination of homology modeling, molecular docking, and long-time molecular dynamics simulation on a lipid bilayer. Our model can reasonably interpret the results of a number of mutation experiments regarding GLP-1R as well as the successful modification to GLP-1 by Liraglutide. Our model is also validated by a recently revealed crystal structure of the extracellular domain of GLP-1R. An activation mechanism of GLP-1R agonists is proposed based on the principal component analysis and normal mode analysis on our predicted GLP-1R structure. Before the complete structure of GLP-1R is determined through experimental means, our model may serve as a valuable reference for characterizing the interactions between GLP-1R and its agonists.
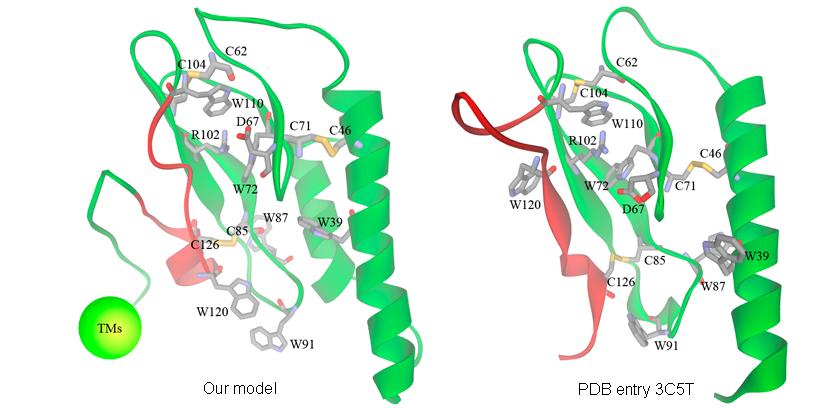
Figure. Our predicted model of rGLP-1R (left) and the crystal structure of hGLP-1R recently revealed by Runge et al. (right). Only the extracellular domain (residues 1-146) on our model is shown in this figure. The only segment apparently different between our model and Runges structure is colored in red.
PROTEINS: Structure, Function, and Bioinformatics 64:1058C1068 (2006)
A Computational Analysis of the Binding Affinities of FKBP12 Inhibitors Using the MM-PB/SA Method
Yong Xu and Renxiao Wang*
The FK506-binding proteins have been targets of pharmaceutical interests over years. We have studied the binding of a set of 12 nonimmunosuppressive small-molecule inhibitors to FKBP12 through molecular dynamics simulations. Each complex was subjected to 1-ns MD simulation conducted in an explicit solvent environment under constant temperature and pressure. The binding free energy of each complex was then computed by the MM-PB/SA method in the AMBER program. Our MM-PB/SA computation produced a good correlation between the experimentally determined and the computed binding free energies with a correlation coefficient (R2) of 0.93 and a standard deviation as low as 0.30 kcal/mol. The vibrational entropy term given by the normal mode analysis was found to be helpful for achieving this correlation. Moreover, an adjustment to one weight factor in the PB/SA model was essential to correct the absolute values of the final binding free energies to a reasonable range. A head-to-head comparison of our MM-PB/SA model with a previously reported Linear Response Approximation (LRA) model suggested that the MM-PB/SA method is more robust in binding affinity prediction for this class of compounds.
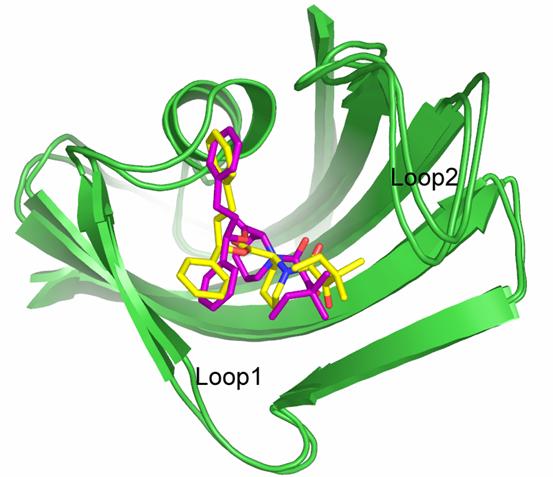
Figure. The crystal structure of the 8-FKBP12 complex superimposed with the final snapshot from the MD trajectory of this complex. Carbon atoms on compound 8 in the crystal structure and the MD snapshot are colored in purple and yellow, respectively.
Copyright ©2007-2021 上海盈赛思信息科技有限公司 网站备案号:沪ICP备2021015625号-2 ![]() 沪公网安备:正在申请中
沪公网安备:正在申请中
Technical Support(技术支持): yingsaisi@foxmail.com